
ABSTRAK
Arma chinensis , serangga predator yang terkenal karena keanekaragaman mangsanya di Asia Timur, efektif dalam mengendalikan hama pertanian dan kehutanan. Namun, setelah memperkenalkan populasi lapangan ke subkultur dalam ruangan, fitur depresi perkawinan sedarah telah muncul dalam populasi ini. Mengklarifikasi mekanisme genetik molekuler dari depresi perkawinan sedarah A. chinensis sangat penting untuk perlindungan populasinya. Dalam penelitian ini, analisis filogenomik mengungkapkan bahwa genus Arma berbagi nenek moyang yang sama dengan Halyomorpha dan Nezara dalam famili Pentatomidae sekitar 63,62 juta tahun yang lalu. Berdasarkan resequencing genom utuh dari tiga generasi perkawinan sedarah berturut-turut dari A. chinensis , kami menyelidiki fitur genom depresi perkawinan sedarah. Kami mengamati akumulasi homozigositas jangka panjang dan variasi ekstrem dalam keanekaragaman nukleotida lintas generasi, yang secara kolektif memengaruhi 111 gen dan beberapa proses biologis, seperti pengikatan DNA spesifik sekuens, organisasi sinaps, dan pengikatan wilayah pengatur transkripsi. Perubahan genomik ini menunjukkan bahwa perkawinan sedarah yang terjadi secara berturut-turut dapat mengganggu fungsi fisiologis normal, yang berpotensi merusak ekspresi gen, sinyal saraf, dan perkembangan organ sensorik. Sebagai kesimpulan, penelitian kami memperjelas posisi evolusi A. chinensis , menyoroti konsekuensi genetik perkawinan sedarah, dan menekankan pentingnya melestarikan keragaman genetik dalam populasi alami untuk kelangsungan hidup dan kemampuan beradaptasi jangka panjang.
1 Pendahuluan
Arma chinensis (ordo: Hemiptera; famili: Pentatomidae; Gambar 1A ) adalah serangga predator yang terkenal karena keanekaragaman mangsanya, yang secara efektif menekan hama dari famili seperti Lepidoptera, Coleoptera, Hymenoptera, dan Hemiptera (Deyu et al. 2012 ; M. Pan et al. 2019 ; H. Wu et al. 2019 ). A. chinensis sebagian besar ditemukan di Asia Timur, termasuk Tiongkok, Semenanjung Korea, Jepang, dan Mongolia (M.-Z. Pan et al. 2022 ; Rider et al. 2002 ), yang menunjukkan kemampuan beradaptasi lingkungan yang luar biasa. Dengan kondisi ekologi yang sesuai, A. chinensis dapat membentuk populasi alami di area yang dilepaskan, sehingga memfasilitasi pengelolaan hama jangka panjang (M.-Z. Pan et al. 2022 ). Oleh karena itu, A. chinensis menonjol sebagai predator bermanfaat yang mampu mengendalikan hama dalam skala luas, secara bersamaan mengurangi bahaya yang terkait dengan pengelolaan hama kimia dan memajukan pengembangan sistem pengendalian hama yang komprehensif (Shen et al. 2018 ). Namun demikian, setelah pengenalan populasi lapangan ke dalam kultur dalam ruangan, fitur depresi perkawinan sedarah, termasuk penurunan tingkat penetasan telur dan peningkatan waktu yang dibutuhkan untuk mencapai dewasa, secara bertahap muncul dalam populasi A. chinensis (Shaoming et al. 2021 ; Zhu et al. 2022 ). Situasi ini menimbulkan hambatan yang cukup besar terhadap perbanyakan dan pemanfaatan pertanian serangga bermanfaat ini, yang menekankan urgensi untuk mengeksplorasi konsekuensi genetiknya.
Secara tradisional, strategi pembiakan sering kali melibatkan berbagai tindakan, termasuk perkawinan sedarah, untuk menghasilkan galur homozigot atau perkawinan sedarah, sehingga memperbaiki sifat-sifat yang diinginkan (Burgarella et al. 2019 ). Namun, perkawinan sedarah yang berkelanjutan selama beberapa generasi, khususnya pada populasi kecil, dapat menyebabkan beberapa konsekuensi genetik. Ini termasuk hilangnya keragaman genetik (Eszterbauer et al. 2015 ), fiksasi alel yang merusak (Assaf et al. 2015 ; Smeds dan Ellegren 2023 ), peningkatan beban genetik (Marsden et al. 2016 ; Wang et al. 2017 ), dan akhirnya, depresi perkawinan sedarah (D. Charlesworth dan Willis 2009 ; Han et al. 2021 ).
Perkawinan sedarah telah diketahui memiliki efek buruk pada serangga (Kuriwada et al. 2010 ; Peng et al. 2015 ; Woldemelak 2024 ). Penelitian terkini tentang depresi perkawinan sedarah pada serangga sebagian besar berfokus pada proses kehidupan dan konsekuensi biologis perkawinan sedarah (Collet et al. 2020 ; Pilakouta et al. 2015 ). Namun, masih kurangnya eksplorasi dasar genetik molekuler dari seluruh genom, termasuk tiga aspek identifikasi gen atau jalur spesifik yang terpengaruh, pemahaman cara kerjanya, dan interaksinya dengan adaptasi pada spesies serangga. Akibatnya, pemeriksaan komprehensif dan menyeluruh dari mekanisme genetik yang mendasari depresi perkawinan sedarah pada serangga sangat penting untuk pemahaman yang lebih bernuansa tentang fenomena ini.
Penelitian ini bertujuan untuk menyelidiki dasar genetik depresi perkawinan sedarah selama pemeliharaan dalam ruangan A. chinensis dengan mengintegrasikan genomik dengan pengurutan ulang genom secara keseluruhan. Pertama, berdasarkan perakitan genom A. chinensis yang dilaporkan (Fu et al. 2024 ), kami melakukan analisis filogenomik untuk menentukan status evolusinya. Selanjutnya, galur uji perkawinan sedarah buatan dibuat dalam kondisi dalam ruangan, diikuti oleh pengurutan ulang genom secara keseluruhan. Hal ini memungkinkan kami untuk menyelidiki dasar-dasar genetik setiap generasi, dengan demikian menjelaskan mekanisme genetik molekuler yang mendorong depresi perkawinan sedarah pada A. chinensis . Dengan memanfaatkan wawasan dari penelitian ini, kami bertujuan untuk menyempurnakan metode untuk meremajakan populasi A. chinensis , mengurangi dampak negatif perkawinan sedarah, dan meningkatkan pembiakan dalam ruangannya untuk aplikasi yang dioptimalkan dalam pengendalian biologis.
2 Bahan dan Metode
2.1 Identifikasi Keluarga Gen, Analisis Filogenomik, dan Analisis Perluasan dan Kontraksi Keluarga Gen
Urutan protein dari A. chinensis , 7 spesies Hemiptera lainnya ( Aphis gossypii , Apolygus lucorum , Cimex lectularius , Halyomorpha halys , Melanaphis sacchari , Nezara viridula , Rhopalosiphum maidis ), dan 1 outgroup ( Drosophila melanogaster ) dianalisis di situs web OrthoVenn3 (Tabel 1 ) (Sun et al. 2023 ). Transkrip terpanjang dipilih untuk mewakili setiap gen dengan beberapa isoform. Urutan protein dari A. chinensis dan 8 spesies lainnya digunakan untuk klasifikasi famili menggunakan OrthoFinder (Emms dan Kelly 2019 ) dengan nilai E 1e-2 dan nilai inflasi 1,5. Klaster ortolog salinan tunggal diselaraskan menggunakan MUSCLE (Edgar 2004 ), dan sekuens yang dilestarikan diekstraksi dan dipangkas dengan trimAI (Capella-Gutiérrez et al. 2009 ). FastTree (Price et al. 2010 ) kemudian digunakan untuk menyimpulkan pohon filogenomik di antara spesies melalui metode kemungkinan maksimum (Felsenstein 1981 ). Analisis kontraksi dan ekspansi keluarga gen dilakukan dengan CAFÉ5 (Mendes et al. 2021 ) di situs web OrthoVenn3. CAFÉ5 menggunakan ambang batas 0,05 dan secara komprehensif menilai ekspansi atau kontraksi signifikan keluarga gen berdasarkan nilai- p seluruh Keluarga , nilai- p Viterbi , dan nilai- p Cut . Perkiraan didasarkan pada waktu fosil ( H. halys — N. viridula hingga 59,15 juta tahun lalu, A. gossypii — A. lucorum hingga 290,05 juta tahun lalu, C. lectularius — H. halys hingga 217,31 juta tahun lalu, R. maidis — N. viridula hingga 290,05 juta tahun lalu, D. melanogaster — R. maidis hingga 361,53 juta tahun lalu, D. melanogaster — A. gossypii hingga 361,53 juta tahun lalu) yang diperoleh dari TimeTree (Kumar et al. 2017 ) menggunakan situs web OrthoVenn3. Keluarga gen yang diperluas dan dikontrak dalam A. chinensis sebagai gen spesifik dibangun untuk analisis pengayaan GO.
| Jenis | dari (default dari NCBI) |
|---|---|
| Halyomorpha halys | FPB_000696795.2 |
| Nezara viridula | GCA_928085145.1 |
| Rhopalosiphum spp. (Rhapsodyne) bunga mawar | Situs web OrthoVenn3 ( https://orthovenn3.bioinfotoolkits.net/ ) |
| Kutu daun | FPB_020184175.1 |
| Melanaphis sakarin | FPB_002803265.2 |
| Apolygus lucorum | GCA_009739505.2 |
| Jamur Tiram | FPB_000648675.2 |
| Lalat buah Drosophila melanogaster | GCA_000001215.4 |
| Arma Cina | GCA_040285775.1 |
2.2 Sampel untuk Resequencing Genom Utuh
Sampel induk A. chinensis dikumpulkan dari ladang di Kota Linyi, Provinsi Shandong, Tiongkok (35°50 LU, 118°38 BT), dan dibudidayakan di dalam ruangan dengan perkawinan sedarah bebas yang berkesinambungan antara generasi yang sama. Suhu pemberian pakan dalam ruangan dijaga pada 26°C ± 1°C, kelembaban relatif 66% RH, dan periode cahaya 16 L: 8 D. Sebanyak 18 individu jantan dewasa (9 F1, 4 F2, dan 5 F3) dari tiga generasi perkawinan sedarah berturut-turut dipilih, dan organ dalam dikeluarkan untuk pengurutan selanjutnya. Sampel-sampel ini digunakan untuk konstruksi pustaka berpasangan sepanjang 150 bp. Genom dari 18 individu ini kemudian diurutkan ulang menggunakan platform Illumina NovaSeq 6000 (Illumina, CA, AS), menghasilkan 189,83 Gb pembacaan bersih.
2.3 Deteksi Variasi Genetik
Pembacaan sekuensing genom secara keseluruhan dipangkas oleh Trimmomatic (v0.39) (Bolger et al. 2014 ) dan dinilai oleh FastQC (v0.12.1). Pembacaan bersih dipetakan ke rakitan genom A. chinensis menggunakan BWA (v0.7.17-r1188) (Li dan Durbin 2009 ). Pembacaan penyelarasan ditandai sebagai terduplikasi, diurutkan, dan diindeks menggunakan SAMtools (v1.20) (Li 2011 ). Deteksi varian dilakukan untuk setiap sampel menggunakan GATK (v3.8.0) dengan parameter default (McKenna et al. 2010 ). BCFtools (v1.9) (Danecek et al. 2021 ) digunakan untuk mengidentifikasi SNP. SNP difilter menggunakan VCFtools (v0.1.16) (∔mac 1—alel-min 2—alel-maks 2—minGQ 30—minQ 300—DP-maks 80) (Viluma et al. 2022 ).
2.4 PCA dan Analisis Filogenomik untuk Populasi A. chinensis
Analisis PCA dilakukan dengan berkas VCF tersaring akhir menggunakan PLINK (v1.90b6.21) (Purcell et al. 2007 ) dan divisualisasikan oleh paket R scatterplot3d (v0.3–44) (Ligges dan Maechler 2003 ). IQ-TREE (v2.2.6) (Nguyen et al. 2015 ) digunakan untuk membangun pohon filogenomik. ModelFinder (Kalyaanamoorthy et al. 2017 ) digunakan untuk pemilihan model, dan model terbaik diperoleh sebagai mtZOA+F + G4, dan kemudian metode kemungkinan maksimum digunakan untuk membangun pohon evolusi menggunakan model terbaik ini. Pohon filogenomik divisualisasikan oleh paket R ggtree (v3.11.2) (Xu et al. 2022 ).
2.5 Rangkaian Deteksi Homozigositas dan Keanekaragaman Nukleotida
Bahasa Indonesia: Mengikuti rekomendasi oleh Meyermans et al. ( 2020 ), pemangkasan ketidakseimbangan hubungan (LD) dan frekuensi alel minor (MAF) tidak dilakukan sebelum mendeteksi run of homozygosity (ROH) (Meyermans et al. 2020 ). Analisis ROH dilakukan oleh PLINK (∔homozyg-window-snp 5—homozyg-kb 5). F ROH didefinisikan sebagai proporsi genom autosomal dalam ROH yang melebihi panjang yang ditentukan (McQuillan et al. 2008 ). Fitur terkait ROH, termasuk distribusi F ROH , jumlah ROH, dan panjang rata-rata ROH, divisualisasikan oleh paket R ggplot2 (v3.5.1) (Ginestet 2011 ). Gen yang dicakup oleh daerah ROH yang melebihi 400 kb khusus untuk generasi F3 A. chinensis sebagai gen spesifik dieksekusi untuk analisis pengayaan GO. Keragaman nukleotida untuk SNP dideteksi menggunakan VCFtools (∔site-pi). Uji-t sampel independen dilakukan menggunakan fungsi t_test dari paket rstatix (v0.7.2) (Kassambara 2023 ). Plot Manhattan dibuat dengan paket ggplot2, berdasarkan sampel acak 1% SNP dari setiap generasi. Plot sankey divisualisasikan menggunakan fungsi sankeyNetwork dari paket networkD3 (v0.4) (Gandrud et al. 2017 ), yang mengilustrasikan transisi SNP antara bin keragaman nukleotida yang berbeda di tiga generasi. Analisis pengayaan fungsional GO dilakukan pada gen yang dicakup oleh situs yang sangat terkonservasi/beragam (π = 0/1) khusus untuk populasi F2 dan F3.
2.6 Analisis Pengayaan Fungsional GO
Beberapa analisis GO dilakukan untuk mengeksplorasi dampak spesifik dari berbagai perubahan genom yang diakibatkan oleh perkawinan sedarah. Beberapa analisis GO disusun dengan tujuan mengeksplorasi dampak spesifik yang disebabkan oleh berbagai jenis perubahan genom dan konsekuensi potensialnya. Basis data organisme untuk A. chinensis dibuat oleh paket R AnnotationForge (v1.45.0) (Carlson et al. 2016 ). Analisis pengayaan fungsional GO dari gen-gen spesifik dilakukan oleh paket R ClusterProfiler (v4.11.1) dengan parameter default (T. Wu et al. 2021 ; Yu et al. 2012 ). Gen-gen spesifik telah didefinisikan dalam Bahan dan Metode sebelumnya. Hasil pengayaan divisualisasikan oleh paket R ggplot2.
3 Hasil
3.1 Sejarah Evolusi dan Analisis Perbandingan A. chinensis
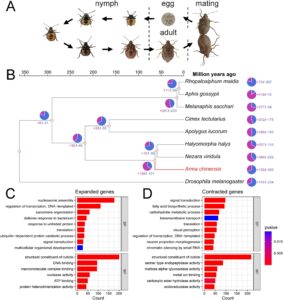
Berdasarkan perakitan genom tingkat kromosom A. chinensis yang dilaporkan oleh Fu et al. (Fu et al. 2024 ), kami selanjutnya menentukan posisi evolusinya. Kami melakukan analisis filogenomik menggunakan sekuens protein dari A. chinensis , 7 spesies Hemiptera lainnya, dan 1 outgroup ( D. melanogaster ) (Tabel 1 ). Semua gen pengkode protein dikelompokkan ke dalam 17.057 ortogrup berdasarkan homologi sekuens. Selain itu, kami mengidentifikasi total 4772 famili gen yang dibagi di antara semua 9 spesies, bersama dengan 402 famili gen spesifik spesies di A. chinensis (Gambar S1 ). Pengayaan GO dari gen-gen ini menunjukkan pengayaan perakitan miofibril, integrasi DNA, pengikatan DNA, perkembangan organisme multiseluler, dan konstituen struktural kutikula (Gambar S1 ).
Pohon filogenomik dibangun menggunakan 1194 ortolog salinan tunggal (Gambar S2 ). Analisis filogenomik mengungkapkan bahwa genus Arma berbagi nenek moyang yang sama dengan Halyomorpha dan Nezara dalam famili Pentatomidae sekitar 63,62 juta tahun yang lalu (Gambar 1B ). Dinamika famili gen mendeteksi 139 dan 309 perluasan dan kontraksi famili gen dalam genom A. chinensis , masing-masing. Analisis pengayaan GO menunjukkan bahwa famili gen yang diperluas dalam A. chinensis dikaitkan dengan konstituen struktural kutikula, perakitan nukleosom, regulasi transkripsi dan DNA-templated, pengikatan DNA, dan pengikatan kompleks makromolekul (Gambar 1C ). Selain itu, analisis menunjukkan bahwa famili gen yang berkontraksi terkait dengan konstituen struktural kutikula, transduksi sinyal, proses biosintesis asam lemak, aktivitas endopeptidase tipe serin, dan proses metabolisme karbohidrat (Gambar 1D ).
3.2 Depresi Perkawinan Sedarah Disertai Akumulasi Rangkaian Panjang Daerah Homozigositas
Untuk menyelidiki mekanisme molekuler yang mendorong depresi perkawinan sedarah pada A. chinensis , resequencing genom secara keseluruhan dilakukan pada 18 orang dewasa dari tiga generasi perkawinan sedarah berturut-turut (9 F1, 4 F2, dan 5 F3) yang dibesarkan di dalam ruangan. Proses di atas menghasilkan 189,83 Gb data berkualitas tinggi (~10,87× cakupan per sampel), yang menghasilkan identifikasi 3.158.715 SNP yang memenuhi syarat setelah penyelarasan dengan genom referensi A. chinensis . Baik analisis filogenomik maupun PCA secara konsisten menunjukkan pola pengelompokan yang lemah, yang mengungkapkan pergeseran genetik populasi kecil yang terkait dengan perkawinan sedarah (Gambar S3–S4 ).
Selanjutnya, fitur ROH dari tiga generasi perkawinan sedarah A. chinensis yang berurutan dideteksi. Tren peningkatan pada F ROH diamati, yang menunjukkan munculnya depresi perkawinan sedarah (Gambar 2A ). Sementara jumlah total daerah ROH tidak menunjukkan tren yang jelas (Gambar 2B ), panjang rata-rata ROH meningkat secara signifikan pada generasi F3 (Gambar 2C ), yang mengarah pada akumulasi panjang total ROH dan kenaikan selanjutnya pada F ROH . Akumulasi ini terutama didorong oleh peningkatan signifikan pada daerah ROH yang panjang (> 400 kb) (Gambar 2D ), yang mencakup 2229 gen yang spesifik untuk generasi F3.
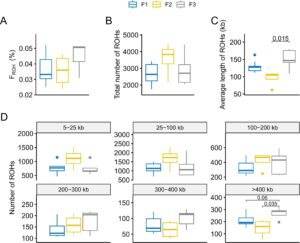
Untuk menilai dampak perkawinan sedarah pada daerah ROH yang panjang ini, kami melakukan analisis pengayaan GO pada gen yang dicakup oleh ROH yang melebihi 400 kb pada generasi F3 (Gambar S5 ). Analisis tersebut mengungkapkan pengayaan yang signifikan dalam proses biologis yang penting, termasuk aktivitas hidrolase, respons seluler terhadap senyawa nitrogen, perkembangan sistem imun, persepsi sensorik, proses efektor imun, regulasi negatif diferensiasi sel, dan generasi gamet jantan. Temuan ini menunjukkan efek genomik dan fenotipik yang luas terkait dengan akumulasi daerah ROH yang panjang akibat perkawinan sedarah.
3.3 Variasi Ekstrim Keanekaragaman Nukleotida ( π ) Akibat Perkawinan Sedarah
Setelah menjalani tiga generasi perkawinan sedarah berturut-turut, pola keragaman nukleotida yang berbeda telah muncul (Gambar 3A,B ). Generasi F1 menunjukkan distribusi keragaman nukleotida yang relatif tersebar, dengan proporsi yang lebih tinggi jatuh antara 0,25 dan 0,75. Sebaliknya, generasi F2 dan F3 menunjukkan puncak yang jelas pada dua ekstrem: π = 0 untuk situs yang sangat terkonservasi dan π = 1 untuk situs yang sangat beragam. Secara khusus, situs ekstrem ( π = 0 dan π = 1) pada generasi F2 sebagian besar mewarisi dari situs non-ekstrem pada generasi F1, dan variasi SNP ini menjadi tetap pada generasi F3 (Gambar 3C ). Di satu sisi, munculnya situs yang sangat beragam menunjukkan potensi untuk kombinasi dan adaptasi genetik baru. Di sisi lain, munculnya situs yang sangat terkonservasi menyiratkan hilangnya keragaman, kemungkinan karena fiksasi alel spesifik dan penyempitan kumpulan genetik berikutnya.
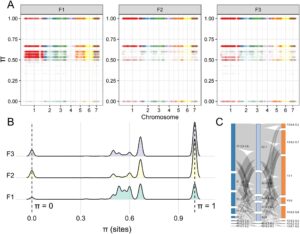
Untuk lebih menjelaskan temuan ini, analisis pengayaan GO dilakukan untuk gen yang tercakup oleh situs ini. Situs yang sangat terkonservasi khusus untuk generasi F2 dan F3 diperkaya dalam proses yang terkait dengan kemotaksis, regulasi ukuran struktur anatomi, kadar hormon, pertumbuhan perkembangan, pengikatan faktor transkripsi, sekresi, perkembangan proyeksi neuron, dan morfogenesis postembrionik (Gambar S6 ). Sebaliknya, situs yang sangat beragam menunjukkan pengayaan dalam proses yang melibatkan komitmen nasib sel, transpor anion, pensinyalan sinaptik, aktivitas faktor transkripsi pengikat DNA, morfogenesis organ sensorik, dan transmisi sinaptik kimia (Gambar S7 ). Ini menunjukkan bahwa sementara pembiakan memperkuat sifat genetik tertentu, itu juga dapat memicu mekanisme kompensasi yang berpotensi mengurangi efek negatif dari depresi bawaan (Zhang et al. 2023 ).
Sebagai kesimpulan, pengamatan kami menyoroti sifat ganda dari proses perkawinan sedarah. Meskipun dapat memperbaiki variasi genetik induk, proses ini secara bersamaan mengurangi keragaman genetik secara keseluruhan dengan memperkuat sifat genetik yang dilestarikan. Analisis pengayaan GO memberikan wawasan tentang proses biologis spesifik yang dipengaruhi oleh perubahan genetik ini, yang menjelaskan interaksi kompleks antara variasi genetik dan konservasi dalam konteks perkawinan sedarah.
3.4 Berbagai Konsekuensi Genom yang Disebabkan oleh Perkawinan Sedarah yang Berturut-turut
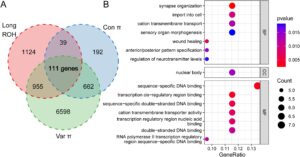
Perkawinan sedarah telah menyebabkan berbagai konsekuensi genomik, termasuk akumulasi daerah Long ROH dan munculnya situs keragaman nukleotida ekstrem. Proses-proses ini secara kolektif telah memengaruhi 111 gen (Gambar 4A ). Analisis pengayaan GO mengungkapkan bahwa gen-gen ini secara signifikan diperkaya dalam fungsi-fungsi yang terkait dengan pengikatan DNA spesifik urutan, organisasi sinaps, regulasi transkripsi, transpor transmembran kation, proses tubuh nuklir, morfogenesis organ sensorik, penyembuhan luka, dan regulasi neurotransmitter (Gambar 4B ), yang menyoroti peran penting mereka dalam berbagai proses biologis dan fungsi molekuler yang berpotensi terganggu oleh efek merugikan dari perkawinan sedarah berturut-turut.
4 Diskusi
Arma chinensis , serangga musuh alami predator dengan kisaran mangsa yang luas, memiliki pengendalian yang efektif terhadap hama pertanian dan kehutanan. Fu et al. ( 2024 ) melaporkan perakitan genom tingkat kromosom berkualitas tinggi untuk A. chinensis , yang mencakup 986 Mb dan mengandung 20.853 gen penyandi protein yang diprediksi (Fu et al. 2024 ). Berdasarkan hal ini, kami selanjutnya menyelidiki evolusi genom dan divergensi A. chinensis . Dengan menggunakan 1194 ortolog salinan tunggal dari 9 spesies serangga, analisis filogenomik mengungkapkan bahwa genus Arma berbagi nenek moyang yang sama dengan Halyomorpha dan Nezara dalam famili Pentatomidae sekitar 63,62 juta tahun yang lalu (Gambar 1B ). Secara keseluruhan, genom A. chinensis menyediakan sumber daya yang berharga untuk analisis hilir.
Penelitian sebelumnya tentang perkawinan sedarah sebagian besar berpusat di sekitar proses kehidupan dan konsekuensi biologis dari perkawinan sedarah. Misalnya, perkawinan sedarah kupu-kupu raja menyebabkan kematian yang lebih tinggi dan umur keturunan yang lebih pendek Mongue et al. ( 2016 ). Di sini kami membuat garis perkawinan sedarah A. chinensis selama tiga generasi berturut-turut untuk menyelidiki konsekuensi tepat perkawinan sedarah pada genom serangga. Hasil kami menunjukkan bahwa perkawinan sedarah memberikan pengaruh yang mendalam pada seluruh genom A. chinensis , mengganggu berbagai proses biologis dan fungsi seluler yang vital untuk perkembangan, diferensiasi, dan pertumbuhan.
Seiring dengan perkembangan generasi perkawinan sedarah, tren peningkatan F ROH mengindikasikan pertumbuhan segmen homozigot akibat perkawinan sedarah (McQuillan et al. 2008 ). Secara spesifik, peningkatan tetap F ROH dari generasi ke generasi dikaitkan dengan peningkatan signifikan panjang rata-rata ROH pada generasi F3 (Gambar 2C ), yang terutama didorong oleh peningkatan signifikan daerah ROH panjang (>400 kb) (Gambar 2D ). Lebih jauh lagi, ada peningkatan nyata baik pada frekuensi situs yang sangat terkonservasi ( π = 0) maupun situs yang sangat beragam ( π = 1) di seluruh genom, yang khususnya terbukti pada populasi F2 dan F3. Transformasi ini mengindikasikan perubahan substansial dalam keragaman nukleotida, yang mencerminkan dampak genetik yang mendalam dari perkawinan sedarah yang berkelanjutan (Ørsted et al. 2019 ; Zhang et al. 2023 ).
Perlu dicatat bahwa kemunculan dan stabilisasi cepat sejumlah situs yang sangat terkonservasi dalam jangka waktu yang singkat menunjukkan potensi hilangnya keragaman genetik, yang dapat membuka jalan bagi efek kemacetan genetik berikutnya (Ali dan Roossinck 2008 ; Roy 2022 ). Munculnya kemacetan populasi akan semakin memperburuk hilangnya keragaman genetik melalui pergeseran genetik yang intensif (B. Charlesworth 2009 ; Grossen et al. 2020 ; Lucena-Perez et al. 2021 ). Pergeseran genetik yang diamati ini menggarisbawahi kerapuhan keragaman genetik, khususnya dalam subpopulasi yang terisolasi secara genetik (Exposito-Alonso et al. 2022 ). Kerapuhan ini menggarisbawahi kebutuhan mendesak untuk aliran genetik eksternal sebagai tindakan mitigasi terhadap potensi efek perkawinan sedarah yang berkepanjangan (Garcia-Cisneros et al. 2016 ; Stevens et al. 2018 ). Kontrol kualitas, yang mencakup pemindahan serangga dewasa yang baru berganti kulit ke kandang serangga dewasa generasi berikutnya, dapat membantu memulihkan koloni serangga sedarah Bagrada hilaris (Ivey et al. 2023 ). Akibatnya, disarankan untuk memperkenalkan populasi eksternal selama pemeliharaan serangga dalam kultur laboratorium untuk mengurangi masalah yang disebabkan oleh depresi perkawinan sedarah (Hedrick dan Garcia-Dorado 2016 ; Johnson et al. 2010 ). Secara khusus, laboratorium harus secara berkala memperkenalkan individu baru dari populasi liar ke dalam kultur mereka untuk meningkatkan keragaman genetik, memantau keragaman genetik secara teratur menggunakan alat genomik seperti resequencing genom utuh, dan mempertahankan ukuran populasi yang lebih besar untuk mengurangi efek negatif perkawinan sedarah.
Munculnya situs yang sangat beragam menunjukkan potensi untuk kombinasi dan adaptasi genetik baru. Sementara pembiakan memperkuat sifat genetik tertentu, hal itu juga dapat memicu mekanisme kompensasi yang berpotensi untuk mengurangi efek negatif dari depresi perkawinan sedarah. Namun, hal ini dapat mengganggu kombinasi genetik adaptif lapangan asli. Selain itu, akumulasi daerah ROH yang panjang (> 400 kb) ditambah dengan berkurangnya keragaman genetik karena perkawinan sedarah dapat semakin membahayakan ketahanan dan kemampuan beradaptasi populasi terhadap gangguan lingkungan. Proses-proses ini secara kolektif memengaruhi 111 gen dalam genom A. chinensis . Analisis pengayaan GO mengungkapkan hubungan yang signifikan dengan berbagai proses dan fungsi biologis, termasuk pengikatan DNA spesifik urutan, organisasi sinaps, regulasi transkripsi, transportasi transmembran kation, proses tubuh nuklir, morfogenesis organ sensorik, penyembuhan luka, regulasi neurotransmitter, antara lain. Dalam penelitian nyamuk baru-baru ini, banyak variasi genom ditentukan oleh pengurutan ulang genom, yang memengaruhi fungsi pencernaan nyamuk, perkembangan, dan kekebalan bawaan (Acharya et al. 2024 ). Temuan ini menggarisbawahi potensi disfungsi biologis yang meluas akibat perkawinan sedarah (Zhang et al. 2023 ).
5 Kesimpulan
Singkatnya, kami menentukan posisi evolusi A. chinensis berdasarkan perakitan genom tingkat kromosomnya (Fu et al. 2024 ). Resequencing genom lengkap dari tiga generasi perkawinan sedarah berturut-turut dari A. chinensis mengungkapkan konsekuensi genetik yang mendalam dari perkawinan sedarah. Temuan ini menyoroti pentingnya melestarikan keragaman genetik, khususnya pada subpopulasi yang terisolasi secara genetik. Wawasan ini dapat menginformasikan upaya konservasi untuk meremajakan populasi A. chinensis , mengurangi dampak negatif perkawinan sedarah, dan menyempurnakan praktik pembiakan dalam ruangan untuk aplikasi pengendalian biologis yang lebih baik.


